



Directrices GEIS 2023 para tumores del estroma gastrointestinal-GIST
Autores
|
César Serrano cserrano@vhio.net , Javier Martín-Broto, […], y en nombre de GEIS (Grupo Español de Investigación en Sarcomas)+10Ver todos los autores y afiliaciones |
ABSTRACTO
El tumor del estroma gastrointestinal (GIST) es la neoplasia maligna de origen mesenquimal más frecuente. Los GIST abarcan un amplio espectro clínico que va desde tumores sin potencial metastásico hasta enfermedades malignas y potencialmente mortales. Las mutaciones de ganancia de función en los receptores tirosina quinasa KIT o PDGFRA son los impulsores cruciales de la mayoría de los GIST, responsables del inicio del tumor y de su evolución a lo largo de todo el curso de la enfermedad. La introducción de inhibidores de la tirosina cinasa dirigidos contra estos receptores ha mejorado sustancialmente los resultados en este cáncer antes quimiorresistente. A día de hoy, hay cinco fármacos aprobados para el tratamiento del GIST: imatinib, sunitinib, regorafenib, ripretinib y avapritinib. Esto, a su vez, representa un éxito para una neoplasia poco frecuente. Durante las dos últimas décadas, el GIST se ha convertido en un modelo paradigmático en cáncer para el trabajo multidisciplinar, dadas las particularidades específicas de la enfermedad en cuanto a biología tumoral y evolución tumoral. A continuación, revisamos la evidencia actualmente disponible para el manejo del GIST. Esta guía de práctica clínica ha sido desarrollada por un panel multidisciplinar de expertos (oncólogo, patólogo, cirujano, biólogo molecular, radiólogo y representante de grupos de defensa de pacientes) del Grupo Español de Investigación en Sarcomas, y está concebida para proporcionar, desde una perspectiva crítica, el abordaje estándar para el diagnóstico, tratamiento y seguimiento.
El tumor del estroma gastrointestinal (GIST), el subtipo de sarcoma más frecuente,1 se origina en el tubo digestivo y se origina principalmente por mutaciones de ganancia de función en los receptores tirosina quinasa (RTK) KIT o PDGFRA.2,3
En las dos últimas décadas, el GIST se ha convertido en un modelo paradigmático para el desarrollo racional y exitoso de agentes molecularmente dirigidos contra el cáncer. Como resultado, cinco inhibidores de la tirosina quinasa (TKI) han sido aprobados para el tratamiento del GIST metastásico: imatinib, sunitinib, regorafenib, ripretinib y avapritinib.4 El beneficio de estos agentes ha dado lugar a un aumento significativo de la supervivencia global (SG), de menos de 12 meses a más de 5 años, 5 que, a su vez, constituye un éxito notable para una neoplasia poco frecuente. En conjunto, estos avances han remodelado el manejo clínico de los pacientes con GIST, desarrollándose actualmente en equipos multidisciplinares con conocimientos entrelazados y complementarios. Dada la creciente complejidad y los recientes avances en este campo, aquí revisamos y actualizamos nuestras guías previas del Grupo Español de Investigación en Sarcomas (GEIS) para el manejo del GIST.6–8
Epidemiología y presentación clínica
La incidencia anual de GIST en España, 1,24 casos/100.000,9 es similar a la registrada en todo el mundo, que oscila entre 1,1 y 1,5/100.000.10 Los GIST se presentan predominantemente en individuos de mediana edad y mayores, con un pico en la séptima década de la vida. Existe una distribución equitativa en todos los grupos sexuales, geográficos, raciales y étnicos. El diagnóstico de GIST suele iniciarse por síntomas inespecíficos como dolor abdominal, hemorragia gastrointestinal u obstrucción. El GIST se origina en el estómago (55%), intestino delgado (31%), colorrectal (6%), esófago (<1%) u órganos extragastrointestinales (<5%).10 En el momento del diagnóstico, aproximadamente el 15% de los pacientes presentan metástasis, que suelen encontrarse en el peritoneo y el hígado.
La gran mayoría de los GIST son esporádicos y carecen de factores etiológicos conocidos. No obstante, algunas condiciones hereditarias predisponen a una mayor probabilidad de GIST(Figure 1) . El síndrome de Carney-Stratakis se asocia a mutaciones inactivadoras de la línea germinal en las subunidades B, C o D de la succinato deshidrogenasa (SDH), que conducen a una presentación precoz de GIST multifocal, con predominio gástrico, crecimiento lento y posibilidad de desarrollar paragangliomas.11 Además, el 7-10% de los pacientes con neurofibromatosis tipo I (NF1) desarrollan GIST asociados a NF1 que típicamente crecen como implantes múltiples a través del intestino delgado.12 Los GIST familiares debidos a mutaciones de la línea germinal de KIT o PDGFRA son extraordinariamente raros 13 y pueden asociarse a hiperpigmentación cutánea, disfagia y trastornos mastocitarios. La derivación de estos pacientes a unidades de consejo genético es muy recomendable. Por último, nosotros y otros autores hemos demostrado una incidencia de 2,5 a 4 veces mayor de segundas neoplasias tras el diagnóstico inicial de GIST,14,15 aunque hasta el momento no existen pruebas suficientes para considerar a estos pacientes como una enfermedad hereditaria; por lo tanto, en general no se recomienda derivarlos, a menos que las características específicas del paciente y de la familia pudieran ser relevantes.

Figura 1. Alteraciones moleculares en GIST esporádicos y familiares. Alteraciones moleculares en GIST esporádicos y familiares.
Los GIST se clasifican en KIT/PDGFRA mutante y KIT/PDGFRA WT. Los GIST WT se dividen en SDH deficientes y SDH competentes según la expresión SDHB IHC. La mayoría de las mutaciones notificadas en GIST son esporádicas, aunque una minoría son mutaciones de línea germinal relacionadas con síndromes específicos o GIST familiares. La prevalencia de las categorías moleculares se expresa en porcentajes.
GIST: tumor del estroma gastrointestinal; IHC: inmunohistoquímica; SDH: succinato deshidrogenasa; WT: tipo salvaje.
Evaluación diagnóstica
Imágenes
El diagnóstico radiológico en los GIST es comparable al realizado en otros tumores del tubo digestivo. Los estudios ecográficos muestran los GIST como lesiones submucosas e hipoecogénicas que, si aumentan de tamaño, pueden desplazar estructuras vecinas y volverse quísticas, necróticas o hemorrágicas. La tomografía computarizada (TC) abdominal es la primera opción para estudiar la localización y la extensión. Los tumores primarios aparecen como masas exoluminales bien circunscritas que pueden mostrar un realce de contraste heterogéneo que representa áreas necróticas-hemorrágicas o degenerativas.16 La TC con contraste y las adquisiciones de imágenes de las fases arterial y portal están indicadas para identificar las lesiones hepáticas hipervasculares subcentimétricas que, de otro modo, pasarían inicialmente desapercibidas, pero que se vuelven hipodensas si responden con el tratamiento TKI de primera línea. La resonancia magnética está indicada en la evaluación del GIST rectal, mientras que la tomografía por emisión de positrones (PET)-CT rara vez se utiliza durante el estudio inicial.17
Biopsia
Una biopsia preoperatoria no es necesaria cuando la evaluación de la lesión primaria se considera sospechosa de GIST y se considera resecable quirúrgicamente. Por el contrario, una biopsia del tumor está indicada bajo un diagnóstico radiológico dudoso y/o en el caso de tumores localmente avanzados que requieran terapia neoadyuvante.17
En la enfermedad localizada, la técnica de elección para el diagnóstico histológico es la ecoendoscopia. La biopsia percutánea guiada por TC sólo puede considerarse si la primera opción no es posible. En este caso, debe prestarse especial atención para evitar la rotura o el derrame del tumor. La biopsia es preferible a la aspiración con aguja fina para obtener material suficiente para realizar un diagnóstico histológico y molecular definitivo.18 En pacientes con enfermedad metastásica accesible, puede preferirse una biopsia guiada por TC o ecografía para abordar el tumor primario.
Características patológicas
Características macroscópicas
Los GIST son de tamaño variable, desde lesiones centimétricas hasta grandes masas tumorales de más de 20 cm. Típicamente, los GIST surgen de la pared del tubo digestivo y se extienden hacia dentro, hacia la mucosa, hacia fuera, hacia la serosa, o en ambas direcciones.19 Con poca frecuencia, pueden ulcerar la mucosa suprayacente. Pueden observarse áreas macroscópicas de necrosis, hemorragia y degeneración quística.19 Mientras que los GIST esporádicos crecen como masas solitarias, diferentes patrones pueden hacer sospechar de entidades particulares, como GIST con deficiencia de SDH si son multinodulares,11 y NF1 o GIST familiares si están presentes múltiples tumores primarios.20 La rotura del tumor es un factor pronóstico adverso independientemente de si tiene lugar antes o durante la cirugía.21
Histología
Pueden distinguirse tres subtipos histológicos: de células fusiformes (77%), epitelioide (8%) y mixto (15%), sin importancia pronóstica.22,23 El tipo epitelioide es más frecuente en el estómago y se asocia a mutaciones del PDGFRA. 22 El recuento mitótico tiene valor pronóstico y debe expresarse como el número de mitosis en un área total de 5 mm2. Deben aplicarse criterios estrictos: los núcleos picnóticos, disquerióticos o apoptóticos no deben considerarse mitosis. Debido a la heterogeneidad intratumoral, la evaluación de las características de estratificación del riesgo, como el recuento mitótico, debe diferirse a la muestra de resección quirúrgica, ya que una biopsia central puede orientar erróneamente su evaluación.
Inmunohistoquímica
Más del 95% de los GIST muestran expresión de KIT (CD117) -tinción citoplasmática, membranosa o perinuclear puntiforme- independientemente del estado mutacional. La tinción fuerte y difusa es el patrón más común presente en el 75% de los casos. Además, los GIST también pueden expresar CD34 (70-90%), actina (20-30%), S-100 (8-10%) y desmina (2-4%).19,23 Una pequeña proporción de GIST (<5%) puede mostrar una expresión débil o nula de KIT, lo que se observa con mayor frecuencia en GIST con mutación PDGFRA24,25 y GIST desdiferenciados. 26 La inmunotinción ANO1/DOG1 es un marcador sensible y específico que es positivo en aproximadamente la mitad de los GIST KIT negativos.27 La inmunohistoquímica PDGFRA también puede ser útil en esta población para predecir la presencia de mutaciones PDGFRA.28
Diagnóstico diferencial
El principal diagnóstico diferencial del GIST de células fusiformes comprende tumores del músculo liso (leiomioma y leiomiosarcoma), fibromatosis (desmoide), schwannoma, tumor maligno de la vaina del nervio periférico, tumor miofibroblástico inflamatorio, tumor fibroso solitario, pólipo fibroide inflamatorio y carcinoma sarcomatoide. El diagnóstico diferencial del GIST epitelioide incluye carcinoma pobremente diferenciado, tumor neuroendocrino, leiomiosarcoma epitelioide, PEComa y melanoma maligno, entre otros.29
| Evaluación diagnóstica: Recomendaciones finales |
- El TAC abdominal con contraste y adquisición de imágenes de las fases arterial y portal es la primera elección para estudiar localización y extensión (IV,B).
- La biopsia preoperatoria es necesaria cuando existe un diagnóstico radiológico dudoso y/o en el caso de tumores localmente avanzados o metastásicos (IV,C).
- El informe patológico debe incluir información para la evaluación del riesgo (localización anatómica, tamaño tumoral, actividad mitótica) y otros factores pronósticos importantes como la rotura tumoral y el estado de los márgenes (Tabla 1) (III,A).
- Un panel básico de inmunohistoquímica (IHC) incluye CD117, DOG1, actina, desmina, S-100 y CD34 (IV,B).
| Tabla 1. Elementos requeridos en el informe patológico de un GIST resecado. |
|
Biología molecular
La determinación del perfil molecular de los GIST es fundamental para adaptar el uso de los agentes dirigidos aprobados contra esta enfermedad. Aproximadamente el 85% de los GIST están causados por mutaciones activadoras mutuamente excluyentes en KIT o PDGFRA. Además, entre el 10% y el 15% de los GIST son de tipo salvaje (WT) para KIT y PDGFRA, y en su patogénesis intervienen otros eventos4 (Figure 1). Las mutaciones en KIT y PDGFRA suelen agruparse en regiones bien conocidas de estas quinasas e incluyen una amplia gama de alteraciones de ganancia de función como deleciones, mutaciones sin sentido, duplicaciones, inserciones y la combinación de éstas.30
Mutaciones KIT
Entre el 75% y el 80% de los GIST albergan mutaciones clonales primarias en KIT, y los exones más comúnmente afectados incluyen el 11, 9, 13 y 17. La mayoría de las mutaciones de KIT se encuentran en el exón 11 (~70%), que codifica el dominio intracelular de yuxtamembrana.4 La mayoría de las alteraciones detectadas en el exón 11 son deleciones que se encuentran predominantemente entre los codones 550 y 579. Menos frecuentemente, las mutaciones missense afectan a los codones 557, 13 y 17. Con menor frecuencia, las mutaciones sin sentido afectan a los codones 557, 559, 560 y 576.31–33 El exón 9 de KIT codifica para el dominio extracelular de unión al ligando, se detecta en el 12-15% de los casos y se asocia con la localización en el intestino delgado y un mayor potencial maligno.30,33 Estas mutaciones consisten comúnmente en duplicaciones de los codones 502 y 503. Por último, los exones 13 y 17 codifican el sitio de unión al ATP de KIT y el bucle de activación, respectivamente. Sólo se producen como sustituciones sin sentido y son bastante infrecuentes como mutaciones primarias (<1%).4,30,33
Las mutaciones primarias en KIT son el evento clonal iniciador presente durante todo el curso de la enfermedad. Por el contrario, las mutaciones secundarias en KIT surgen tras la presión selectiva ejercida por los TKI y están asociadas a la resistencia en hasta el 90% de los pacientes con GIST. Las mutaciones secundarias de KIT se agrupan en el bolsillo de unión al ATP (exones 13 y 14) y en el bucle de activación (exones 17 y 18)34,35 (Figure 2).

Figura 2. GIST con mutaciones en KIT y PDGFRA.
Las mutaciones primarias de KIT afectan comúnmente a los exones 9 y 11, y con menor frecuencia a los exones 17 y 18, mientras que las mutaciones secundarias de KIT se agrupan en el bolsillo de unión al ATP y en el bucle de activación; se muestran los aminoácidos afectados con mayor frecuencia. Las mutaciones primarias en PDGFRA surgen de regiones similares. La prevalencia de las categorías moleculares de las mutaciones primarias de KIT y PDGFRA se expresa en porcentajes.
Ex, exón; GIST, tumor del estroma gastrointestinal.
Mutaciones PDGFRA
La frecuencia estimada de mutaciones en PDGFRA oscila entre el 5% y el 10% y se asocia a GIST de localización gástrica y morfología .36 Las mutaciones se encuentran principalmente en los dominios yuxta membrana (0,7%) y quinasa (6%) codificados por los exones 12 y 18, respectivamente. La sustitución D842V en el exón 18 es la alteración más frecuente del PDGFRA (65-75% de los casos de PDGFRA mutado). Las mutaciones en el exón 14 son extremadamente infrecuentes (0,1%)36 (Figure 2).
GIST KIT/PDGFRA tipo salvaje
El GIST KIT/PDGFRA WT incluye una mezcolanza de alteraciones moleculares y fenotipos clínicos resultantes que es necesario identificar para predecir el curso de la enfermedad, descubrir síndromes hereditarios y, si está indicado, adaptar adecuadamente tratamientos específicos.
GIST con deficiencia de succinato deshidrogenasa
l complejo proteico SDH está compuesto por cuatro subunidades: SDHA, SDHB, SDHC y SDHD. La pérdida de cualquiera de estas subunidades por mutaciones o silenciamiento epigenético conduce a la inestabilidad de todo el complejo SDH, lo que provoca la degradación de SDHB y la consiguiente pérdida de su expresión. Por esta razón, la inmunohistoquímica para SDHB es un marcador diagnóstico útil en la identificación de este subgrupo..37 En general, se estima que más del 80% de los GIST adultos KIT/PDGFRA WT son deficientes en SDH.38
Clínicamente, estos tumores tienden a aparecer en la población pediátrica (sin predominio de sexo) y en la adultez joven (con predominio femenino) y están restringidos al estómago.11 Los GIST con deficiencia de SDH tienen rasgos morfológicos característicos, incluyendo afectación multinodular de la pared gástrica y ocasional invasión de ganglios linfáticos. Microscópicamente, estos GIST presentan morfología epitelioide, fuerte expresión de KIT (CD117) y pérdida de expresión de SDHB en las células tumorales. Aunque el espectro clínico puede ser amplio, la progresión suele ser lenta incluso después de la diseminación, y muchos pacientes pueden vivir años con metástasis.37–39
GIST competente en succinato deshidrogenasa
Menos del 20% de los GIST KIT/PDGFRA WT CD117-positivos son SDH competentes y, por tanto, conservan la expresión de SDHB. Este subgrupo de tumores se caracteriza por otras anomalías genéticas que conducen a la sobreactivación de la vía de la proteína cinasa activada por mitógenos, como mutaciones de pérdida de función de NF1, mutaciones de ganancia de función de BRAF V600E o reordenamientos cromosómicos que implican al receptor neurotrófico de la tirosina cinasa (NTRK).12,38–40
Los GIST con mutación NF1- y BRAF se producen casi exclusivamente a lo largo del intestino delgado. Sin embargo, mientras que los GIST con mutación BRAF son tumores aislados, los GIST con mutación NF1 se presentan más comúnmente como una enfermedad multifocal o multinodular. En este caso, debe excluirse un síndrome hereditario de NF1 no reconocido previamente..41 Actualmente no existen características morfológicas o de IHC específicas, y el diagnóstico final sólo se realiza mediante paneles de secuenciación de nueva generación (NGS).
Por último, estudios exhaustivos de perfiles genómicos han identificado una serie de fusiones génicas en GIST con KIT/PDGFRA WT que implican un reordenamiento cromosómico que afecta a NTRK3, lo que representa una alteración procesable. 40 Sin embargo, estos casos parecen ser excepcionalmente raros. 42 Dada su rareza y la alta tasa de falsos positivos para la tinción de NTRK en tumores no fusionados con NTRK, el cribado rutinario en GIST para anomalías de NTRK no es apropiado, y los paneles de NGS pueden usarse sólo si la evaluación conjunta eleva la sospecha diagnóstica.40
Análisis de secuenciación de Sanger con falsos negativos
La sensibilidad de la secuenciación Sanger rutinaria es del 20-25%. Sin embargo, es posible que la frecuencia alélica de mutaciones heterocigotas de KIT o PDGFRA sea menor en muestras tumorales que contengan una proporción anormalmente elevada de tejido normal o infiltrado inmunitario. En nuestra experiencia GEIS, el 40-50% de los GIST KIT/PDGFRA WT por secuenciación Sanger resultaron ser KIT- o PDGFRA-mutantes cuando se analizaron con NGS, que tiene una mayor sensibilidad debido a su capacidad para detectar mutaciones en fracciones alélicas más bajas del 1-5%. 43
| Biología molecular: Recomendaciones finales |
- Se recomienda encarecidamente el análisis molecular sistemático durante el estudio diagnóstico de todos los GIST, dada la importante información predictiva y pronóstica que proporciona. El análisis genético también confirma el diagnóstico de GIST en tumores CD117/DOG-1 negativos (II,A).
- Es aconsejable centralizar el análisis mutacional en laboratorios inscritos en un programa externo de garantía de calidad y con experiencia en GIST (IV,C).
- Se recomienda el uso de paneles de NGS en GIST KIT/PDGFRA WT sin deficiencia de SDH (III,A).
- Se recomienda la recogida de tejido fresco/congelado ya que se pueden realizar nuevas evaluaciones de patología molecular en fases posteriores en interés de los pacientes (V,C).
Estratificación del riesgo en GIST localizados
La evaluación del riesgo de recaída en el GIST primario es primordial no sólo para proporcionar información pronóstica al intentar determinar los factores de riesgo, sino también para estimar el beneficio potencial del imatinib adyuvante.
Sistemas de estratificación del riesgo
El primer índice propuesto (National Institute of Health – NIH Consensus o índice de Fletcher et al.) consideraba el tamaño y el recuento mitótico por 50 campos de alta potencia (HPF) como las variables con valor pronóstico independiente.22 Miettinen y Lasota propusieron un índice más complejo que incluía la localización del tumor primario tras observar una menor tasa de recurrencia en el GIST gástrico en comparación con otras localizaciones. Así, los criterios de riesgo del Instituto de Patología de las Fuerzas Armadas (AFIP)/Miettinen y Lasota incorporaron la localización anatómica al tamaño tumoral y al recuento mitótico.44 Últimamente, los criterios modificados por consenso de los NIH incorporaron la rotura tumoral a estos tres factores pronósticos, y fueron la base para la estimación del riesgo en los ensayos aleatorizados que estudiaban el imatinib adyuvante en GIST localizados45 (Table 2).. Es importante destacar que Miettinen consideró un área total de 5 mm2 en 50 HPF caracterizados por el uso de diferentes componentes ópticos, mientras que en la práctica 50 HPF suelen corresponder a un área total de 10 mm2. Por lo tanto, si se utiliza la clasificación de riesgos de Miettinen, el número de HPF equivalentes a una superficie de 5 mm2 debe calcularse en función de los parámetros específicos del microscopio.
Tabla 2. Directrices para la evaluación del riesgo de GIST primario.
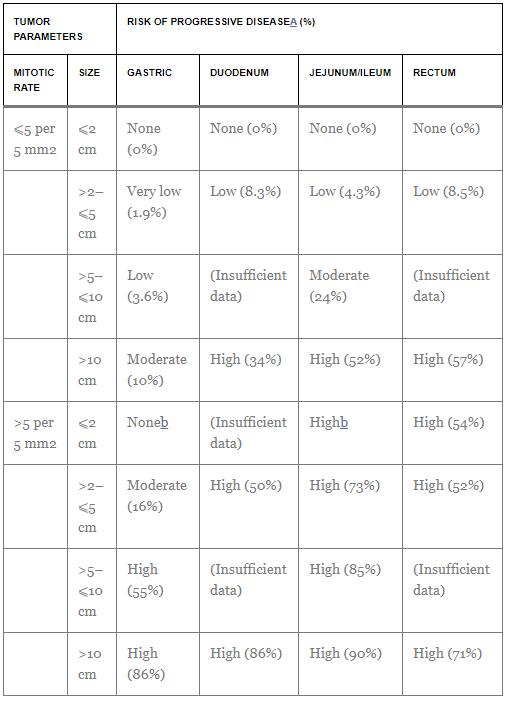
Fuente: Adaptado con permiso de Miettinen y Lasota. Copyright 2006 por Elsevier.
a
Definido como metástasis o muerte relacionada con el tumor.
b
Indica un número reducido de casos.
GIST, tumor del estroma gastrointestinal.
La casuística del grupo GEIS demostró que la clasificación de la AFIP, a diferencia de los criterios de riesgo de los NIH, presentaba significación estadística para distinguir los grupos de riesgo bajo, intermedio y alto.32 Otras clasificaciones de riesgo alternativas conllevan las limitaciones de las variables categorizadas. Para superar este obstáculo, Joensuu et al. propusieron una clasificación de riesgo basada en el modelado no lineal del recuento y el tamaño mitóticos, analizándolos como variables continuas. La precisión de esta predicción se realizó en un registro de base poblacional, y la generación de mapas térmicos es superior a la de otros modelos de clasificación del riesgo cuando se comparó el área bajo la curva. Las directrices de la Sociedad Europea de Oncología Médica son proclives a utilizar los mapas de calor como la mejor herramienta para ofrecer una estimación del riesgo. El imatinib adyuvante suele recomendarse para aquellos casos con una probabilidad de recurrencia superior al 40% utilizando el modelo de mapas de calor.46
Características moleculares predictivas del pronóstico de riesgo
Existen pruebas que indican que el tipo y la localización de la mutación influyen en el riesgo de recurrencia. Nosotros descubrimos que las deleciones que afectan a los codones 557 y/o 558 en el exón 11 de KIT (a partir de ahora, nos referiremos a dichas deleciones como «mutación crítica») tienen un mayor riesgo de recurrencia en los primeros 3-4 años tras la cirugía,32 lo que se confirmó en series más amplias.47,48 Un estudio retrospectivo posterior de la red Conticanet que resumía 1.056 casos de GIST localizados descubrió que los casos gástricos de riesgo intermedio que albergaban «mutaciones críticas» tenían un peor pronóstico significativo e independiente.49 Las mutaciones dentro de PDGFRA mostraron una tendencia hacia un mejor pronóstico.
| Estratificación del riesgo: Recomendaciones finales |
- Recomendamos los criterios de riesgo modificados por consenso de los NIH, ya que han sido ampliamente utilizados para la recomendación de imatinib adyuvante (III,A).
- Las mutaciones de tipo deleción que afectan a los codones 557 y 558 confieren riesgo de recurrencia independientemente de su clasificación previa, según diferentes series retrospectivas (III,A).
Tratamiento de la enfermedad localizada
Cirugía
La resección quirúrgica completa es el tratamiento estándar para el GIST localizado. El objetivo es una cirugía R0 con extirpación completa del tumor, incluida una pseudocápsula intacta. El margen de resección macroscópica recomendado es de 1 cm. La resección tumoral debe realizarse cuidadosamente para evitar la rotura del tumor.21,50 Las superficies peritoneal y hepática deben examinarse cuidadosamente para excluir la diseminación tumoral. La linfadenectomía es innecesaria excepto en los GIST deficientes en SDH, o si se detecta afectación macroscópica de los ganglios linfáticos. Generalmente se acepta la resección segmentaria del intestino y el estómago (resección no anatómica/resección en cuña), evitando así procedimientos agresivos con extirpación innecesaria de tejido no afectado. Los criterios radiológicos de irresecabilidad incluyen la infiltración del tronco celíaco, la arteria mesentérica superior o la arteria mesentérica a la vena porta. Ocasionalmente, la resección en bloque puede ser considerada; sin embargo, la resección multivisceral debe ser evitada y la consulta multidisciplinaria con un equipo experto en GIST es altamente recomendada.51,52
La resección R1 de un tumor GIST no implica un mayor riesgo de recurrencia ni una peor supervivencia, mientras que una resección macroscópica incompleta (R2) se asocia a un peor pronóstico.53,54 La reintervención tras la cirugía R1 no está bien definida y podría ofrecerse si no implica mayores riesgos o consecuencias funcionales.
La resección laparoscópica debe seguir los mismos principios de la cirugía abierta, con el mismo objetivo de lograr una resección R0.55,56 En general, la resección laparoscópica en GIST no se recomienda en tumores >10 cm debido a un alto riesgo de ruptura tumoral. Puede considerarse en GIST <5 cm en localizaciones anatómicas favorables como la curvatura mayor, el fundus y la cara anterior gástrica. La extracción de la pieza quirúrgica debe realizarse en bolsa para evitar la diseminación locorregional. No obstante, la indicación de resección laparoscópica debe acordarse caso por caso tras una evaluación multidisciplinar por equipos con amplia experiencia en cirugía laparoscópica.
GIST pequeño < 2 cm
Los GIST submucosos esofagogástricos o duodenales < 2 cm se tratan principalmente con ecografía endoscópica. Si es posible realizar una biopsia, debe hacerse el diagnóstico de GIST. Si se confirma, la resección endoscópica con escisión completa y sin rotura tumoral puede ser una alternativa aceptable a las resecciones laparoscópicas o abiertas para minimizar la morbilidad. Como alternativa, los pacientes pueden optar por someterse a vigilancia activa. Asimismo, si una biopsia no es factible o el material es inadecuado, la primera opción suele ser la vigilancia activa, aunque los pacientes pueden optar por la resección quirúrgica o endoscópica. El seguimiento inicial con endoscopia puede realizarse cada 3 meses y prolongarse en ausencia de crecimiento tumoral.17
Tratamiento adyuvante con imatinib
Aunque la resección quirúrgica completa es factible en la mayoría de los GIST localizados, la recidiva metastásica se produce en aproximadamente el 40% de los pacientes.46 Tres ensayos clínicos aleatorizados de fase III evaluaron el papel del imatinib adyuvante para prevenir la recidiva de la enfermedad y mejorar la SG. Los estudios ACOSOG Z900157 y SSGX-VIII/AIO58 mostraron una mejora de la supervivencia libre de recaída (SLR) tras 1 y 3 años, respectivamente, de imatinib adyuvante 400 mg diarios. Además, el ensayo SSGX-VIII/AIO demostró un aumento de la SG con 3 años de administración de imatinib en comparación con 1 año en pacientes de alto riesgo, según los criterios de riesgo modificados por los NIH. Últimamente, el estudio de fase III EORTC 62024 aleatorizó a pacientes de riesgo intermedio y alto a 2 años de imatinib frente a observación.59 A pesar del impacto manifiesto del imatinib en la SLR, no hubo diferencias en la SG ni en la supervivencia libre de fracaso del imatinib (SLFI) -un criterio de valoración innovador que capta el tiempo transcurrido hasta el desarrollo de resistencia secundaria al imatinib en el contexto metastásico-, aunque se observó una tendencia no significativa hacia la mejora de la SLFI en los pacientes de alto riesgo.
Un seguimiento más reciente a 10 años del ensayo SSGX-VIII/AIO confirmó el beneficio a largo plazo de 3 años de imatinib adyuvante, mostrando unas tasas de SG a 10 años del 79% frente al 65% en el grupo de 12 meses.60 A la vista de estos resultados, la comunidad científica está de acuerdo en recomendar 3 años de tratamiento adyuvante con imatinib 400 mg diarios en pacientes de alto riesgo con mutaciones sensibles a imatinib en KIT o PDGFRA.
Quedan pendientes dos cuestiones importantes. En primer lugar, la duración del imatinib adyuvante. Recientemente, el ensayo clínico de un solo brazo PERSIST-5, de fase II, informó de una SSR estimada a 5 años del 90%, superior a los datos históricos, aunque sólo 46 de los 91 pacientes inscritos completaron los 5 años de imatinib adyuvante.61 El ensayo de fase III SSG XXII (NCT02413736), actualmente en curso, aleatoriza a pacientes con GIST de alto riesgo resecados quirúrgicamente a 3 frente a 5 años de imatinib adyuvante. La segunda cuestión se refiere al beneficio potencial del imatinib adyuvante en situaciones particulares, entre las que destacan los pacientes de riesgo intermedio con mutaciones primarias de KIT que afectan a los codones 557 y/o 558. 32,48,49 Aunque los determinantes moleculares del riesgo no se incluyen en el consenso internacional, basándonos en las sólidas pruebas comunicadas por otros y por nosotros, recomendamos el imatinib adyuvante en este subconjunto de pacientes.
| Imatinib adyuvante en situaciones especiales |
- La rotura tumoral, independientemente de que se produzca de forma espontánea o durante el procedimiento quirúrgico, se asocia a un riesgo de recaída cercano al 100%.21 Por lo tanto, estos casos deben considerarse metastásicos y tratarse con imatinib en consecuencia. Sin embargo, la duración total del imatinib puede discutirse individualmente en situaciones especiales como la microperforación tumoral.
- La sensibilidad específica de algunos genotipos primarios de KIT y PDGFRA debe considerarse siempre cuidadosamente para la indicación de imatinib adyuvante. Por ejemplo, el beneficio de la dosis estándar de 400 mg también se ha demostrado retrospectivamente en pacientes con GIST con mutación del exón 9 de KIT,62 un subconjunto de GIST que se beneficia de una dosis más alta de imatinib (véase más adelante). Sin embargo, los GIST con la mutación primaria PDGFRA D842V y todos los GIST WT para KIT y PDGFRA son, por definición, insensibles al imatinib y, por tanto, el imatinib adyuvante no está indicado.
- La cirugía R1, por sí misma, no constituye un criterio a ponderar en un uso potencial de imatinib adyuvante.
Tratamiento neoadyuvante con imatinib
Una estrategia de tratamiento basada en imatinib de primera línea seguida de cirugía puede estar indicada ocasionalmente en pacientes con GIST con enfermedad localmente avanzada. Es obligatoria la discusión previa en una junta tumoral multidisciplinar. El objetivo final es facilitar un procedimiento quirúrgico R0. Por lo tanto, este enfoque puede intentarse en aquellos GIST que son inicialmente resecables, pero a expensas de una cirugía mutilante.63 Ejemplos típicos de estos escenarios son las masas GIST en la unión gastroesofágica, el duodeno o el recto. También pueden considerarse otras localizaciones, sobre todo en casos con alto riesgo de rotura tumoral, es decir, masas necróticas de >10 cm. El perfil molecular antes de iniciar el tratamiento es fundamental, ya que orienta la dosificación o desaconseja el uso de imatinib.
La evaluación precoz de la respuesta, es decir, después de 4 semanas de iniciado el tratamiento, es muy recomendable, ya que un tratamiento con imatinib infructuoso podría impedir una intervención quirúrgica que de otro modo podría haberse llevado a cabo. Aunque un TAC es suficiente para la evaluación de la respuesta, un TEP/TC puede ser más ventajoso para medir la eficacia del imatinib en un corto periodo de tiempo.64 La duración recomendada del imatinib neoadyuvante no puede basarse en criterios objetivos. Sin embargo, se estima que la cirugía podría realizarse entre 6 y 12 meses después del inicio del imatinib, ya que en este intervalo de tiempo se espera una respuesta máxima y un riesgo mínimo de resistencia secundaria.65 El imatinib puede suspenderse 24 h antes de la cirugía y reiniciarse una vez confirmada la tolerancia oral.
El tratamiento adyuvante con imatinib puede estar indicado una vez finalizada la cirugía. Para ello, se tomará un recuento mitótico de la biopsia tumoral obtenida antes del tratamiento neoadyuvante. Asimismo, el tamaño y la localización del tumor se tomarán del TAC basal. Si está indicado, la duración total del tratamiento preoperatorio y postoperatorio con imatinib deberá sumar el total de 3 años de duración de un tratamiento adyuvante convencional.
| Gestión de la enfermedad localizada: Recomendaciones finales |
1.- El tratamiento estándar de los GIST localizados es una resección quirúrgica completa (III,A).
2.- El tratamiento adyuvante con imatinib 400 mg diarios está indicado en pacientes con GIST de alto riesgo de recaída portadores de mutaciones sensibles a imatinib en KIT o PDGFRA (I,A). Imatinib 400 mg diarios es una opción para pacientes con mutaciones en el exón 9 de KIT (IV,C).
3.- El perfil molecular de KIT y PDGFRA es obligatorio antes del inicio de imatinib adyuvante (II,A).
4.- El tratamiento con imatinib neoadyuvante puede considerarse en una junta tumoral multidisciplinar en pacientes con GIST con enfermedad localmente avanzada en los que la reducción del tumor puede facilitar el procedimiento quirúrgico (III,A).
Tratamiento de la enfermedad metastásica
Tratamiento de primera línea con imatinib
Dosis y eficacia
La dosis estándar de imatinib de 400 mg al día66 se estableció a partir de dos ensayos aleatorizados de fase III en GIST metastásicos con inmunotinción positiva para CD117 (EORTC 62005 y NASG S0033)..67,68 En ambos ensayos, las dosis diarias de 400 frente a 800 mg fueron comparables, obteniéndose una tasa de beneficio clínico (respuesta completa, respuesta parcial y enfermedad estable) de ~90%. La supervivencia libre de progresión (SLP) en el ensayo EORTC favoreció a la dosis de 800 mg, con una SLP a 2 años del 52% frente al 44% (HR 0,78), lo que se confirmó en un metanálisis posterior..69 No obstante, dado que esto no se tradujo en una ventaja de supervivencia y que las dosis más bajas se toleraron mejor, la dosis estándar recomendada es de 400 mg diarios.
Valor predictivo del genotipado de KIT y PDGFRA
Las tasas de respuesta objetiva para KIT exón 11, mutantes exón 9 y GISTs WT fueron del 72%, 38% y 28%, respectivamente, con una mediana de tiempo hasta la progresión (mTTP) de 25, 17 y 12,8 meses, respectivamente.70–73 Un metaanálisis posterior confirmó que sólo los GIST mutantes del exón 9 de KIT se benefician de imatinib 400 mg dos veces al día, reduciendo así en un 42% el riesgo de progresión y en un 31% el riesgo de muerte.69 Los datos actuales son insuficientes para proporcionar cifras claras para los exones 13 o 17 de KIT, por lo que se recomienda inicialmente imatinib 400 mg.
Según datos de laboratorio y datos clínicos dispersos, la mutación PDGFRA más común en GIST, D842V, se considera completamente resistente al imatinib..36,74 En cambio, otras mutaciones en PDGFRA que no implican la sustitución D842V parecen ser sensibles.
| Cuestiones prácticas sobre el imatinib de primera línea en pacientes metastásicos |
|
| Tratamiento para pacientes con progresión de la enfermedad tras el fracaso de imatinib |
| Aumento de la dosis de imatinib |
Si el cumplimiento es correcto, debe cambiarse la terapia sistémica. Una opción a considerar es aumentar la dosis a 400 mg dos veces al día. Un total del 30% de los pacientes que pasaron de 400 a 800 mg en los ensayos EORTC 62005 y NASG S003367,78 alcanzaron el control de la enfermedad. Aunque el mTTP fue modesto, 81 días, el 18% de los pacientes permanecieron libres de progresión un año después de la transición. Los pacientes con mutación del exón 9 de KIT parecen beneficiarse especialmente de este enfoque, mientras que es más limitado en los mutantes del exón 11 de KIT.79–81 La incidencia de anemia y astenia aumenta significativamente con esta dosis; por lo tanto, se requiere un seguimiento estricto.67,68
| Segunda línea: Sunitinib |
El inhibidor multicinasa sunitinib es una alternativa igualmente válida al imatinib a dosis altas tras la progresión a imatinib 400 mg. El sunitinib mostró una mejoría del mTTP de 1,5 a 6,3 meses en comparación con el placebo.82 La dosis recomendada es de 50 mg por vía oral una vez al día durante 4 semanas seguidas de un periodo de descanso de 2 semanas. Un ensayo posterior de fase II de un solo brazo con una dosis diaria continua de 37,5 mg mostró una actividad comparable y una mejor tolerabilidad, por lo que constituye una alternativa válida..83 Los efectos secundarios más frecuentes fueron astenia, toxicidad cutánea, diarrea, hipertensión e hipotiroidismo. Algunos antecedentes moleculares específicos se benefician especialmente del tratamiento con sunitinib: mutación primaria del exón 9 de KIT, GIST con deficiencia de SDH y mutaciones secundarias en los exones 13 y 14 de KIT.11,84,85
El sunitinib sigue siendo el tratamiento estándar de segunda línea tras los resultados del ensayo INTRIGUE, en el que el sunitinib y el ripretinib mostraron resultados comparables, con una SLP de 8,3 y 8,0 meses, respectivamente.86
| Tercera línea: Regorafenib |
El inhibidor de la multicinasa regorafenib está aprobado en dosis de 160 mg diarios, 3 semanas sí, 1 semana no, como tercera línea de tratamiento según los resultados del ensayo GRID de fase III87 , que mostró una SLP de 4,8 meses frente a 0,9 en el brazo placebo. Las toxicidades más frecuentes son hipertensión, reacción cutánea mano-pie y diarrea. A diferencia del sunitinib, el regorafenib parece ser más activo contra las mutaciones secundarias de KIT localizadas en el bucle de activación (exones 17 y 18).35
Regorafenib sigue siendo el tratamiento estándar de tercera línea tras los resultados del ensayo VOYAGER, en el que regorafenib y avapritinib mostraron resultados comparables, con una SLP de 5,6 y 4,2 meses, respectivamente.88
| Cuarta línea y más allá |
El ripretinib es un TKI de tipo II que antagoniza tanto el dominio de yuxta membrana como el bucle de activación de KIT y PDGFRA. El ensayo de fase III INVICTUS demostró que ripretinib 150 mg diarios era superior a placebo tras la progresión a todos los tratamientos estándar, mostrando una SLP de 6,3 meses frente a 1,0 mes.89 El perfil de toxicidad de ripretinib es comparable al de imatinib, aunque con una mayor incidencia de alopecia y síndrome mano-pie leve.
Pazopanib 90 y cabozantinib, 91 entre otros TKI multiobjetivo,39 han mostrado cierta actividad interesante en pacientes resistentes a imatinib en ensayos clínicos de fase II, aunque faltan ensayos aleatorizados amplios y no están aprobados para el tratamiento de GIST. Sin embargo, podrían utilizarse como uso compasivo en pacientes seleccionados tras la progresión a todos los tratamientos estándar. Por último, la supresión continua de TKI después de la progresión a un agente dado parece mejorar los resultados de los pacientes metastásicos con GIST, como se ha demostrado con la reintroducción de imatinib92 y el mantenimiento de sunitinib o avapritinib después de la progresión. 93,94 Por lo tanto, dada la rápida progresión de los pacientes con GIST refractarios a TKI,89 recomendamos mantener o reintroducir un TKI previo mientras haya una alternativa terapéutica disponible.
| Pacientes con GIST con mutación PDGFRA D842V |
La sustitución del ácido aspártico por valina en el codón 842 del exón 18 del PDGFRA (D842V) se produce en el ~5% de todos los GIST y se sabe que es resistente a todos los agentes terapéuticos.36,74 La actividad del avapritinib, un TKI de tipo I, se estudió en el ensayo de fase I NAVIGATOR en 56 GIST con mutación D842V, incluidos 11 TKI naïve. La tasa de respuesta global fue del 91%, la tasa de beneficio clínico del 98% y la mediana de SLP de 34 meses, lo que constituye una actividad sin precedentes en este subconjunto molecular de GIST.95,96 Las toxicidades más frecuentes son náuseas, fatiga, anemia, diarrea y edema, y también un aumento característico de los efectos cognitivos en el 37% de los pacientes que requiere una monitorización estricta.97
| La actividad de todos los agentes aprobados para el tratamiento de GIST se resume en la Table 3. |
Tabla 3. Actividad actualizada de los inhibidores de la tirosina quinasa aprobados en GIST avanzado/metastásico.
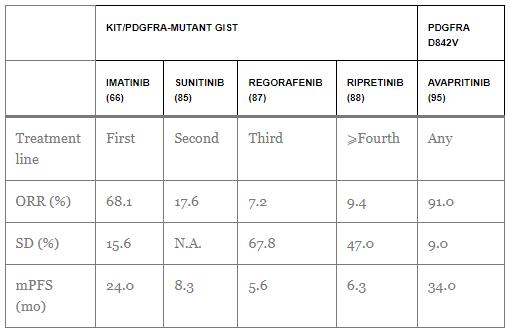
GIST: tumor del estroma gastrointestinal; SSPm: mediana de supervivencia libre de progresión; N.A.: no disponible; ORR: tasa de respuesta global; MS: enfermedad estable (al menos 12 semanas).
| Tratamiento local del GIST metastásico |
Varias series retrospectivas, incluida la nuestra, han demostrado sistemáticamente que la cirugía citorreductora dirigida a la enfermedad R0/R1 tras la respuesta inicial al imatinib se asocia a una mejor supervivencia a largo plazo.98,99 Sin embargo, la resección incompleta, incluida la cirugía citorreductora, no parece prolongar la supervivencia.98,100 La ausencia de datos desaconseja el uso combinado de quimioterapia intraperitoneal hipertérmica (HIPEC). En conjunto, y a pesar de su carácter retrospectivo y probablemente sesgado, estos trabajos apoyan que pacientes seleccionados puedan someterse a cirugía citorreductora tras la respuesta inicial a imatinib y si la enfermedad metastásica se considera resecable. Dada su naturaleza metastásica, es importante evitar procedimientos mutilantes y mantener el imatinib después.
Si bien todos estos estudios no recomiendan la cirugía en pacientes con progresión multifocal, coinciden en que la resección de la progresión unifocal/limitada puede mejorar la SLP. .98,100,101 Esta decisión debe tomarse de forma individual tras debatirse en un comité multidisciplinar de tumores. Aparentemente, este enfoque beneficia principalmente a los pacientes en tratamiento con imatinib. Sin embargo, puede considerarse en líneas posteriores en función del tiempo de administración del fármaco. Al igual que en los pacientes con cirugía citorreductora, se debe continuar con el mismo TKI tras la resección. Por último, la evidencia para otros abordajes como la embolización o la radiofrecuencia es escasa, por lo que debe priorizarse la cirugía.
| Imatinib para la enfermedad metastásica: Recomendaciones finales |
| El genotipo es obligatorio para tratar a los pacientes con GIST avanzado/metastásico (II,A). |
| Imatinib 400 mg diarios es la dosis recomendada en primera línea (I,A). |
| Imatinib 400 mg cada 12 h es la dosis recomendada para GIST con mutación del exón 9 de KIT (II,A). |
| No está claro si imatinib debe ser la primera línea en GIST KIT/PDGFRA WT (IV,C). |
| La cirugía de citorreducción con el objetivo de una cirugía R0/R1 puede ser considerada en pacientes seleccionados después de la respuesta inicial a imatinib (IV,B). |
| Enfermedad resistente al imatinib: Recomendaciones finales |
| Confirmar la adherencia al tratamiento y descartar interacciones farmacológicas en el momento de la progresión a cualquier TKI (III,B). |
| Tras el fracaso de imatinib, el tratamiento estándar de segunda línea es sunitinib 50 mg diarios 4/2 (I,A) o 37,5 mg de forma continua (III,C). |
| Antes del sunitinib, se puede considerar el aumento de la dosis de imatinib a 400 mg dos veces al día, especialmente en pacientes con GIST mutante en el exón 9 de KIT (III,B). |
| Los tratamientos estándar de tercera y cuarta línea son, respectivamente, regorafenib 160 mg diarios 3/1 (I,A) y ripretinib 150 mg una vez al día (I,A). |
| Avapritinib 300 mg diarios es el único tratamiento eficaz disponible para el GIST con mutación PDGFRA D842V y debería introducirse, si es posible, como primera línea (III,A). |
| El mantenimiento de la presión de TKI mejora los resultados y se aconseja mientras no se disponga de una opción terapéutica alternativa (III,B). |
| La cirugía de la progresión unifocal/limitada puede considerarse tras discusión en una junta tumoral multidisciplinar (IV,C). |
| Evaluación y seguimiento de la respuesta |
| Enfermedad resecable localizada |
No existen estudios que analicen la eficacia de las diferentes estrategias de seguimiento. La mayoría de las recomendaciones aceptadas abogan por un ajuste al riesgo de recurrencia con el tiempo en función de los factores de riesgo conocidos.17 Sugerimos los siguientes calendarios según cada grupo de riesgo durante 10 años :46
|
| Enfermedad irresecable o metastásica |
El seguimiento debe realizarse cada 3 meses desde el inicio y puede prolongarse hasta cada 6 meses si se obtiene respuesta, especialmente si ésta se mantiene más allá de los 5 años. Deben tenerse en cuenta los criterios RECIST 1.1 modificado y Choi para evitar la confusión con la pseudoprogresión.
| Conclusiones y perspectivas |
| Desde la última actualización de las directrices GEIS para GIST en 2017, las autoridades reguladoras han aprobado dos nuevos TKI, ripretinib y avapritinib, para el tratamiento de GIST metastásico. Dos ensayos aleatorizados de fase III posteriores han ayudado a consolidar el sunitinib y el regorafenib como segunda y tercera línea de tratamiento estándar, respectivamente. Dado el continuo conocimiento de la biología del GIST y el alto potencial para desarrollar ensayos aleatorizados internacionales, es probable que el campo terapéutico se vea sacudido en los próximos años. En este sentido, animamos a tratar a los pacientes con GIST en cualquier fase de los ensayos clínicos para impulsar el desarrollo de fármacos. |
| En los próximos años se aclararán otros dos aspectos del tratamiento de los GIST: En primer lugar, si 5 años de imatinib adyuvante es superior a 3 años que es el estándar actual en segundo lugar, el papel del ADN tumoral circulante en la toma de decisiones de tratamiento, particularmente en GIST metastásico. Por último, queremos destacar que, a pesar de todo este éxito terapéutico, el GIST es un tumor poco frecuente. Como tal, la evidencia colectiva ha demostrado sistemáticamente que el tratamiento de estos pacientes en centros de referencia de sarcomas mejoraba sus resultados. Por lo tanto, estamos a favor de un diálogo continuo entre los médicos de los pacientes de origen y las instituciones expertas en sarcomas para que los pacientes con GIST sean derivados en su mejor interés. |
| Agradecimientos |
Agradecemos al Grupo Español de Investigación en Sarcomas (GEIS) su apoyo administrativo.

| texto integro y original |
| https://journals.sagepub.com/doi/10.1177/17588359231192388 |
traducción y enmaquetado
«punto de encuentro y de información de los pacientes de gist»